

INTRODUCTION
The Epstein-Barr virus (EBV) infection correlates with numerous human ailments, encompassing infectious mononucleosis (IM), as well as a spectrum of malignancies. The discovery of the EBV in the 1960s by Yvonne Barr, Bert Achong, and Sir Anthony Epstein initiated a scientific quest to understand the virus's consequences in illnesses including infectious mononucleosis and its possible correlation with cancer. The scientific community became more and more interested in the creation of an EBV vaccine as the late 20th century went on (Esau, 2017). Preventive steps became even more important when Neoplasms such as Burkitt and Hodgkin lymphoma, gastric, and nasopharyngeal carcinoma manifest subsequent to the infection, alongside various lymphoproliferative disorders (Niedobitek, Meru, & Delecluse, 2001). The incidence rate and the gravity of these conditions emphasize the potential societal health advantages of an EBV vaccination (Maple et al., 2022). Early efforts to develop vaccines were fraught with difficulties, and there was little chance of producing strong and long-lasting immune responses. Subunit vaccines and inactivated viral particles were the main focus of the early experiments, but efficacy and safety issues forced more improvement (Vashishtha & Kumar, 2021). A new era in vaccination technology was brought about by developments in immunology, genetics, and molecular biology that allowed for the creation of more potent EBV vaccines in the twenty-first century. In the pursuit of an effective vaccination, recombinant DNA technology and viral vector platforms gained importance (Koff et al., 2013). More accurate and effective vaccine candidates have been developed as a result of a greater understanding of the intricate immunological response to EBV, including the discovery of viral antigens and immune evasion mechanisms. Numerous EBV vaccine candidates have entered clinical trials recently, and their safety and immunogenicity have shown encouraging results. Trials in phases I and II have yielded important information about vaccination schedules, doses, and possible side effects (Escalante, Mutsvunguma, Muniraju, Rodriguez, & Ogembo, 2022). Notwithstanding notable advancements, issues including sustained effectiveness, cross-protective immunity, and worldwide accessibility continue to exist. The scientific community is still working together to find solutions to these problems, developing new strategies and improving on already-existing vaccine candidates. The creation of the EBV vaccine illustrates the scientific community's tenacity in the face of complex viral obstacles. There is still optimism that an efficient and safe EBV vaccine may be developed, providing a guard against the related disorders, as research and clinical trials move forward (Neto, Sidney, Grifoni, & Sette, 2023).
Even though EBV has a high illness burden, preventive vaccinations are still lacking, even though the virus was discovered and isolated more than 50 years ago (Escalante et al., 2022). Since the viral major envelope glycoprotein 350/220 (gp350) is the main target for neutralizing antibodies in naturally infected persons, efforts to build a vaccine have centered on this protein. In animal challenge models like the cottontop tamarin, early iterations of gp350-based vaccinations were shown to produce protective immunity against EBV-mediated lymphomas. More recently, a reduction in infection in a rhesus macaque model utilizing rhesus macaque lymphocryptovirus, an EBV homolog, has been shown (Zhong et al., 2022). Several prospective preventive vaccinations against EBV have been tested in humans; the only known phase II experiment using recombinant soluble gp350 with an AS04 adjuvant was for an EBV preventive vaccination; the frequency of IM in individuals who tested negative for EBV was reduced by 78% with this vaccine, but it was not able to stop the spread of primary infection, which has dampened enthusiasm for its further development (Sokal et al., 2007). EBV infiltrates B cells by interacting with its primary receptor, complement receptor 2 (CR2/CD21), through viral gp350 or, alternatively, through CR1 (CD35). The heterotrimeric viral glycoprotein complex, gH/gL/gp42, acts as a co-receptor on B cells, binding to HLA class II molecules (Smith, Coleman, Gewurz, & Rochford, 2020). Concurrently, integrins are engaged as main receptors by heterodimeric gH/gL and BMRF2, which promotes epithelial cell infection. The degree of virus neutralization varies depending on the kind of targeted cell and the specificity of the antibody, even while inhibition of any of these viral glycoproteins by antibodies or gene disruption effectively prevents or severely limits viral cell entrance (Kombe, Zahid, Mohammed, Shi, & Jin, 2021). When applied to B cells infected with EBV, the murine monoclonal antibody 72A1 demonstrates strong neutralizing activity. According to Tanner et al. (2015), the predicted epitope on gp350 strongly corresponds with the assumed binding area of CR2. This alignment points to a neutralization process made possible by the antibody, possibly by interfering with the CR2 binding site and preventing viral attachment and subsequent infection.
Presently, there is no medicinal remedy available for EBV, and neither are there any preventive or therapeutic vaccines. Several initiatives in vaccine development have targeted specific antigenic proteins, notably gp350, given its status as the primary target for naturally occurring neutralizing antibodies and its prevalence on the virus and in infected cells (Kanekiyo et al., 2015). Clinical trials, including a phase 2 trial utilizing an EBV vaccine derived from the gp350 protein, have been conducted (Zhao et al., 2018). While many vaccines primarily target gp350, specific antigens have been pinpointed for their pivotal roles in evading the immune system. These antigens encompass BGLF5, BZLF1, BNLF2a, BILF1, EBNA1, EBNA2, among others (Kanekiyo et al., 2015). If these antigens are evaluated in future clinical studies, their possible incorporation into a preventative vaccine may improve efficacy. Vaccination, which aims to present antigenic elements to the immune system in order to elicit adaptive immunological responses, has shown itself to be a successful therapeutic strategy in the prevention and treatment of a variety of microbial illnesses.
The ability of the virus to secrete proteins with antigenic properties has paved the way for predicting antibody epitopes through in silico methods, marking a crucial stride in vaccine development (Díaz-Dinamarca et al., 2022). This epitopic prediction, made feasible by immunoinformatics methods, significantly contributes to the rapid, reliable, and efficient creation of peptide-based vaccines. These methods involve identifying linear (continuous) or non-linear (non-continuous) epitopes for both B and T cells, aiding in the in-silico design of vaccines without the exhaustive costs and resources typically associated with immunological experiments. As a result, immunoinformatics prediction of potential B and T-cell epitopes for viral antigens is a prominent approach in peptide vaccine development. Additionally, tools for predicting T-cell epitopes help identify allele-specific peptides, which increases the likelihood of selecting and characterizing potential vaccine candidates (Mir et al., 2022). These methods have been successfully applied in the prediction of vaccines against a variety of viruses, including HPV, Ebola, HIV, Nipah, Zika, and others (Olotu & Soliman, 2021). Additionally, immunoinformatic methods have been utilized to forecast antibody epitopes.
Comprehensive B and T-cell epitope predictions of important glycoprotein proteins gp30/22, which mediate attachment to B cells through complement receptor 2 (CR2/CD21), were made in this study. This work has significant implications for the development of highly efficacious preventive vaccinations. Additionally, by evaluating each possible T-cell epitope's intensity of binding to the human MHC-I molecule, HLA-A02:01, the study was able to estimate how immunogenic it would be. Since this specific allele (HLA-A02) is commonly found in a variety of populations and geographical areas, this inquiry will concentrate on the 02 haplotype. The frequencies of MHC-I HLA-B35, MHC-II DRB113, and HLA-DRB115 molecules in human populations have also been emphasized in earlier research. As part of our current study, we analyzed the glycoprotein gp30/22 of the Herpes Simplex Virus (HSV) to find possible immunogenic areas (Sievers et al., 2002).
The reaction of the immune system to a pathogen can significantly vary among different populations due to the influence of host genetic polymorphisms. Within the human genome, the HLA (Human Leukocyte Antigen) genes exhibit the highest degree of polymorphism (Tamouza, Krishnamoorthy, & Leboyer, 2021). This diversity poses a substantial challenge in vaccine development and population coverage because each allele within this genetic group interacts with a specific set of peptides. To address this complexity, most HLA-I alleles have been categorized based on their ability to bind similar peptides. This classification system encompasses over 80% of the HLA-A and HLA-B alleles, aiding in understanding peptide interactions within these genetic variations. Both classes I and II MHC (Major Histocompatibility Complex) molecules serve in presenting T-cell epitopes, which are recognized by distinct subsets of T-cells: CD8 and CD4, respectively, indicating the existence of CD8 and CD4 T-cell epitopes (Neto et al., 2023). Upon encountering their corresponding epitopes, CD8 T-cells transform into cytotoxic T lymphocytes (CTLs), while primed CD4 T-cells differentiate into helper (Th) or regulatory (T-reg) T-cells. MHC-I molecules primarily present peptides generated from intracellular regions, which are commonly recognized by CD8 CTLs (Kervevan & Chakrabarti, 2021). On the other hand, MHC-II molecules present peptides mainly derived from extracellular regions, which are predominantly recognized by CD4 helper T lymphocytes (HTLs), as previously mentioned. Reports suggest that epitopes recognized by CD8 cytotoxic CTLs possess high immunogenicity (Olotu & Soliman, 2021). Inducing a strong immunological response has been linked to their incorporation into vaccination formulations. The CD4 T cell subclass's epitopes also have important augmentative functions in vaccines, affecting the duration and potency of the immune response. Our goal was to identify a likely contender for the creation of a vaccine. We also carried out a thorough genome-wide search to find the best medication target location. Next, we used computational techniques to mimic the inhibition of this target location with a predicted inhibitor chemical. This study's main objective was to provide the framework for upcoming lab-based initiatives focused on developing an all-encompassing approach to treating and preventing herpes virus infection.

MATERIALS AND METHODS
The research methodology employed in developing the strategy against the Epstein-Barr Virus is succinctly depicted in Figure 1. The diagram provides an overview of a vaccination approach targeting the T41 Glycoprotein 350/220 for vaccine formulation. The experimental procedures were executed using a computing platform, specifically a Dell Inspiron [2021 Dell Inspiron 14 5000 2-in-1], featuring an Intel Core i7 processor, 16GB DDR4 RAM, and a 512GB storage capacity. The operating system employed during the experiments was Windows XP Professional Edition. The study made use of both online and offline resources, including the internet. Through the use of bioinformatics techniques, vaccines intended to recognize specific B- and T-cell epitopic peptides isolated from Glycoprotein 350/220 were created. The distinct T41 B-cell and T-cell epitope proteins with the highest antigenic relevance for Epstein-Barr virus subtypes were found in this study using in silico analysis. Antigenicity, stability, and peptide length for particular B and T-cell epitopes within the glycoprotein protein were taken into account as selection criteria. The NCBI database included the complete viral proteome of the strains of Epstein-Barr virus. Several databases and bioinformatics techniques were used to assess the diverse immunological frameworks of conserved T41 sequences. Sequences of amino acids were obtained from the NCBI database. The VaxiJen server and the Kolaskar and Tongaonkar technique were used to determine the antigenic nature of the Glycoprotein 350/220 sequence. After a large number of B and T-cell epitopes were analyzed, the vaccination effectiveness of one effective peptide from each B and T-cell epitope was assessed based on their antigenic determinants. Two similar B-cell epitopes were found after using B-cell epitope prediction servers to analyze the T41 conserved non-structural protein. ABCPred and the Immune Epitope Database were utilized to perform a thorough examination of the amino acid sequences pertaining to B and T-cell epitopes.
Extraction of Sequence Information
The UniProt Knowledge Base (UniProtKB) database provided the Epstein-Barr Virus T41 Glycoprotein 350/220 amino acid sequences in FASTA format (Chang et al., 1998). As a comprehensive database of protein sequences and their corresponding annotations, UniProtKB acts as a focal point for gathering functional information on proteins while guaranteeing accuracy, consistency, and thorough annotation. After being acquired, the sequences were examined for solvent exposure, surface accessibility, flexibility, antigenic qualities, and MHC class I binding sites.
Determining the Protein with Optimal Antigenicity
The subsequent step entailed submitting the proteins to the VaxiJen v2.0 Server, utilizing default parameters, to predict potent antigens and subunit vaccines and identify the protein with the highest antigenic potential (Obaidullah et al., 2021). The target organism designated for analysis was a virus, and a standard sequence format was provided for the examination. Following this, all the antigenic proteins, along with their respective scores, underwent filtration using Excel. For a more thorough investigation, a singular antigenic protein displaying the highest antigenicity values was chosen.
Exploring the Primary and Secondary Structural Landscape of Proteins
The chosen protein's physiological and chemical properties underwent analysis using both the ProtParam tool from the Expasy server (Chakma et al., 2023) and a self-optimized prediction method with alignment (SOPMA) (Chakma et al., 2023). With the predefined settings in ProtParam, various characteristics of the protein were determined, encompassing its theoretical isoelectric point (pI), molecular weight, amino acid composition, grand average hydropathicity (GRAVY), estimated half-life, extinction coefficient, instability index, and aliphatic index. Furthermore, SOPMA provided predictions for properties such as solvent accessibility, transmembrane helices, globular regions, bend regions, random coil regions, and coiled-coil regions. This comprehensive analysis offered valuable insights into the structural and functional attributes of the protein, enhancing our understanding of its overall nature.
Preliminary Steps in Identifying Epstein-Barr Virus T-Cell Epitopes
In this context, NetCTL-1.2, a web-based system specifically designed for predicting human CTL epitopes within any given protein, was utilized (Larsen et al., 2007). It generated an overall score by taking into account factors such as proteasomal cleavage, TAP transport efficiency, and MHC class I affinity predictions. For our present study, we set the threshold value for epitope identification at 0.5, ensuring a balance between sensitivity (0.89) and specificity (0.94). Additionally, an additional resource tool was utilized to forecast multiple crucial factors for every peptide that was chosen, which is found in the Immune Epitope Database (IEDB). According to Donnes and Kohlbacher (2005), they included the proteasomal cleavage score, processing score, MHC-1 binding score, and TAP score (Transporter Associated with Antigen Processing). The SMM (Stabilized Matrix Method) algorithm was used to calculate these scores, which provide important information about each peptide's potential for antigenicity.
Estimating Epitope Stability
The conservation of particular epitopes was predicted using the IEDB analysis resource. In this case, conservation refers to the proportion of protein sequences where the epitope is present at or above a given identity level. The online Java tool known as the epitope conservation calculator was utilized to provide information on the degree of conservation of these epitopes among various protein sequences (Fleri et al., 2017).
Population Inclusivity Calculation
The IEDB population coverage tool was employed to calculate the population coverage for each epitope (Fleri et al., 2017). This tool relies on MHC binding and/or T cell restriction data to estimate population coverage through an online interface. Its primary objective is to determine the percentage of individuals expected to mount a response to a specific set of epitopes, considering their known MHC restrictions. Three important data points were provided by the program for each individual population coverage assessment: expected population coverage, population recognition of HLA combinations, and recognition of 90% of the population (PC90). All epitopes and the corresponding MHC-I molecules were entered into the tool to carry out these computations, and the population coverage metrics were then ascertained.
Analyzing Potential Allergic Responses
To forecast the allergenicity of the epitope intended for vaccine development, we utilized the online tool known as AllerHunter. This service assesses allergenicity by integrating the guidelines established by the Food and Agriculture Organization (FAO) and the World Health Organization (WHO) with the use of support vector machines (SVM) in pairwise sequence similarity analysis. AllerHunter is a helpful tool for allergen cross-reactivity prediction since it can accurately identify both allergenic and non-allergic components with a high degree of specificity (Krutz et al., 2023).
Development of 3D Epitope Spatial Model
To conduct docking simulation studies, we incorporated the highly conserved TLAGPRSKY epitope into the PEP-FOLD server. PEP-FOLD is a de novo approach specifically designed to predict peptide structures based on amino acid sequences (Thevenet et al., 2012). Its methodology relies on utilizing a structural alphabet (SA) that characterizes the structural conformations of four consecutive amino acid residues. This approach associates the predicted sequence of SA letters with both a greedy algorithm and a coarse-grained force field. As a result, the PEP-FOLD server generated five proposed 3D structures. Subsequently, we selected the most suitable model from these structures for the purpose of analyzing its interactions with HLAs.
Computational Docking Exploration
Molecular docking plays a pivotal role in the field of computational drug design, aiming to predict the preferred spatial arrangement of a ligand when interacting with the binding site of a receptor molecule. The strength of this interaction between the ligand and the receptor is quantified using the experimentally determined inhibition constant, denoted as Kd. In our study, a docking analysis was carried out to validate the binding interaction between HLA molecules and the identified epitope, utilizing AutoDock Vina. The binding energy of the receptor-ligand interaction can be quantified by Eq. 1:
DGbind = DGcomplex - (DGligand - DGreceptor)
This relationship between DG and Kd is shown by Eq. 2
DGbind = -RT ln Keq = - RT ln Kd
Molecular docking was performed to evaluate the binding affinity of selected ligands with a target protein, with the objective of identifying the most promising inhibitors. Throughout the docking procedure, specific parameters were configured as follows: dummy atoms were utilized to represent the docking site, the placement phase employed the Triangle Matcher algorithm, receptor refinement was conducted in a rigid manner, both initial and final scoring utilized the London DG scoring function, and a total of 20 poses were retained for each compound to explore ligand interactions with the selected residues within the active site. To determine the most suitable conformation or pose of the inhibitor within the active site of the target protein, the S-score or docking score was utilized as a key criterion (Yang, Chen, & Zhang, 2022).
Prognosticating B-cell Epitope Targets
In forecasting the B-cell epitope, we utilized IEDB's B-cell epitope prediction algorithms to predict linear B-cell epitopes within the provided highly immunogenic protein sequence. Key factors considered for B-cell epitope prediction included flexibility, antigenicity, surface accessibility, hydrophilicity, and linear epitope properties. Our analysis involved the application of various algorithms sourced from the IEDB analysis resource, including Karplus and Schulz for flexibility prediction, Kolaskar and Tongaonkar for antigenicity assessment, Emini for surface accessibility estimation, Parker for hydrophilicity prediction, and Bepipred for linear epitope prediction (Singh, Malik, & Raina, 2021). Additionally, Chou and Fasman's beta-turn prediction tool was applied, guided by wet lab experiments that highlighted the presence of antigenic elements within beta-turn regions of the protein.
RESULTS
Discerning the Protein with Supreme Antigenic Properties
The search for structural and non-structural proteins of Human Herpesvirus yielded a total of 1214 matches. All proteins underwent evaluation through the VaxiJen server, which produced an overall score for each protein sequence, reflecting their potential to elicit an immune response. Notably, the protein sequence identified by the UniProtKB accession number Q99700 attained the highest score of 0.5797 in the VaxiJen analysis among all queried proteins. This particular protein is the glycoprotein T41 glycoprotein 350/220 of the Epstein Barr virus, comprising 877 amino acids. As a result, it has been chosen for additional examination in the current research.

Evaluating Both Primary and Secondary Structure Configurations
The functionality of a protein is intricately linked to its structural characteristics. The ProtParam server analyses the protein sequence, deriving parameters crucial for determining protein stability and function. Similarly, the secondary structural features of a protein offer insights into its functional attributes. Results generated by ProtParam indicate an Instability Index (II) of 42.22, an aliphatic index of 61.94, and a negative GRAVY (grand average hydropathy) of -0.353 for the protein. SOPMA was employed to compute the secondary structural features, revealing that the protein is predominantly comprised of random coils at 62.71%. Alpha Helix and Extended Strands constitute 10.49% and 22.35% of the protein, respectively. Additionally, Beta turns make up 4.45% of the structure. The calculated parameters from both tools are presented in Table 2; Table 1 respectively. The secondary structure plot is visually represented in Figure 2.
Table 1
Distinct Physicochemical Characteristics of Epstein and Glycoproteins T41 and 350/220.
Table 2
SOPMA Assessment of Secondary Structures in Epstein and Glycoproteins T41, 350/220.
Recognition and Mapping of T-Cell Epitopes
Ensuring consistent predictions of CTL epitopes holds significant importance in the rational design of vaccines, notably by minimizing the experimental effort required in wet labs for epitope identification. In this study, the NetCTL-1.2 web-based server (Arfat et al., 2023) was employed for predicting human CTL epitopes within a given protein. The overall score, integrating predictions of proteasomal cleavage, TAP transport efficiency, and MHC class I affinity, guided epitope identification with a set threshold value of 0.75. This selection criterion, with a sensitivity of 0.89 and specificity of 0.94, informed the choice of 7 epitopes for subsequent dry lab experimentation. For MHC-I binding prediction, the Stabilized Matrix Base Method (SMM) (Singh et al., 2021). was utilized to calculate IC50 values representing peptide binding to MHC-I molecules. The peptide length, standardized at 9 amino acids, was applied uniformly for both frequent and non-frequent alleles during prediction. Alleles exhibiting a binding affinity with IC50 values less than 200 nm were further considered. Additionally, an IEDB analysis resource tool was integrated into the study to predict proteasomal cleavage score, TAP score, processing score, and MHC-I binding score for each selected peptide, utilizing the SMM method (Zhang et al., 2008). This comprehensive approach enhances the precision of epitope selection for potential inclusion in vaccine development efforts Table 3.
Calculating Reach Across Demographics
The population coverage for each epitope was determined using the IEDB population coverage tool, which relies on MHC binding and/or T cell restriction data. This tool predicts the fraction of individuals likely to respond to a given set of epitopes with known MHC restrictions. For each epitope, the tool computed the predicted population coverage, HLA combinations recognized by the population, and HLA combinations recognized by 90% of the population (PC90). The population coverage region was chosen prior to submission, and the epitopes and matching MHC-I molecules were supplied. The investigation concentrated on the best MHC-I binders that were found for every epitope. The proportion of people in particular regions who may be sensitive to the query epitopes was used to compute the population coverage. Examining the immunogenetic landscape across several European nations reveals intriguing disparities in Major Histocompatibility Complex class I (MHC-I) coverage and average hit scores. Austria, boasting an MHC-I coverage of 85.17% and an average hit of 9.73, aligns with a robust immune profile. The Czech Republic closely follows suit with an MHC-I coverage of 86.08% and an average hit of 9.74, reflecting a similar immunological resilience. England, with a commendable MHC-I coverage of 87.98% and an average hit of 10.44, showcases a heightened immune responsiveness. A broader perspective on Europe, encompassing diverse nations, exhibits an overall MHC-I coverage of 83.66% and an average hit of 9.32, depicting a collective yet varied immunogenetic makeup. Ireland emerges with notable figures, recording an MHC-I coverage of 87.14% and an average hit of 10.21, highlighting its immunological robustness. In contrast, the Gulf countries, notably Saudi Arabia, exhibit the lowest MHC-I coverage, signalling a potential vulnerability. This information serves as a pivotal foundation for comprehending the differential responsiveness of diverse populations to specific epitopes, thereby informing and optimizing the strategic formulation of vaccines (Figure 3).
Table 3
Optimal 7 T-Cell Epitopes: Examining MHC-1 Affinities, Processing Scores, and Conservancy

Assessment of Allergenic Potential
The assessment of allergenicity for the proposed vaccine epitope was conducted using the online server AllerHunter. This server employs a combinational approach, incorporating the Food and Agriculture Organization (FAO)/World Health Organization (WHO) allergenicity assessment proposal and support vector machines (SVM)-pairwise sequence similarity. AllerHunter is designed to predict both allergens and non-allergens with high specificity, making it a valuable tool for predicting allergen cross-reactivity. By leveraging this comprehensive methodology, AllerHunter enhances the accuracy of allergenicity predictions, taking into account multiple factors crucial for assessing the potential allergic response to the proposed epitope in the context of vaccine development. This information aids researchers in making informed decisions regarding the safety and efficacy of the epitope in consideration for vaccine design (Muh, Tong, & Tammi, 2009). ATNLFLLEL," is classified as a "PROBABLE NON-ALLERGEN" based on the prediction from the AllerHunter server. The nearest protein with the UniProtKB accession number P13547, to which the sequence belongs, is defined as a non-allergen. This information suggests that, according to the AllerHunter prediction and the associated protein in UniProtKB, the given sequence is likely not an allergen.
Optimization of T-Cell Epitope Choice
It appears that the epitope 'ATNLFLLEL' has been identified as a more suitable vaccine candidate among the five primarily selected epitopes. This selection is based on several factors, including overall epitope conservancy, population coverage, and the affinity for the highest number of HLA molecules. These considerations collectively contribute to the favourable assessment of 'ATNLFLLEL' as a potential candidate for vaccine development.
Figure 4
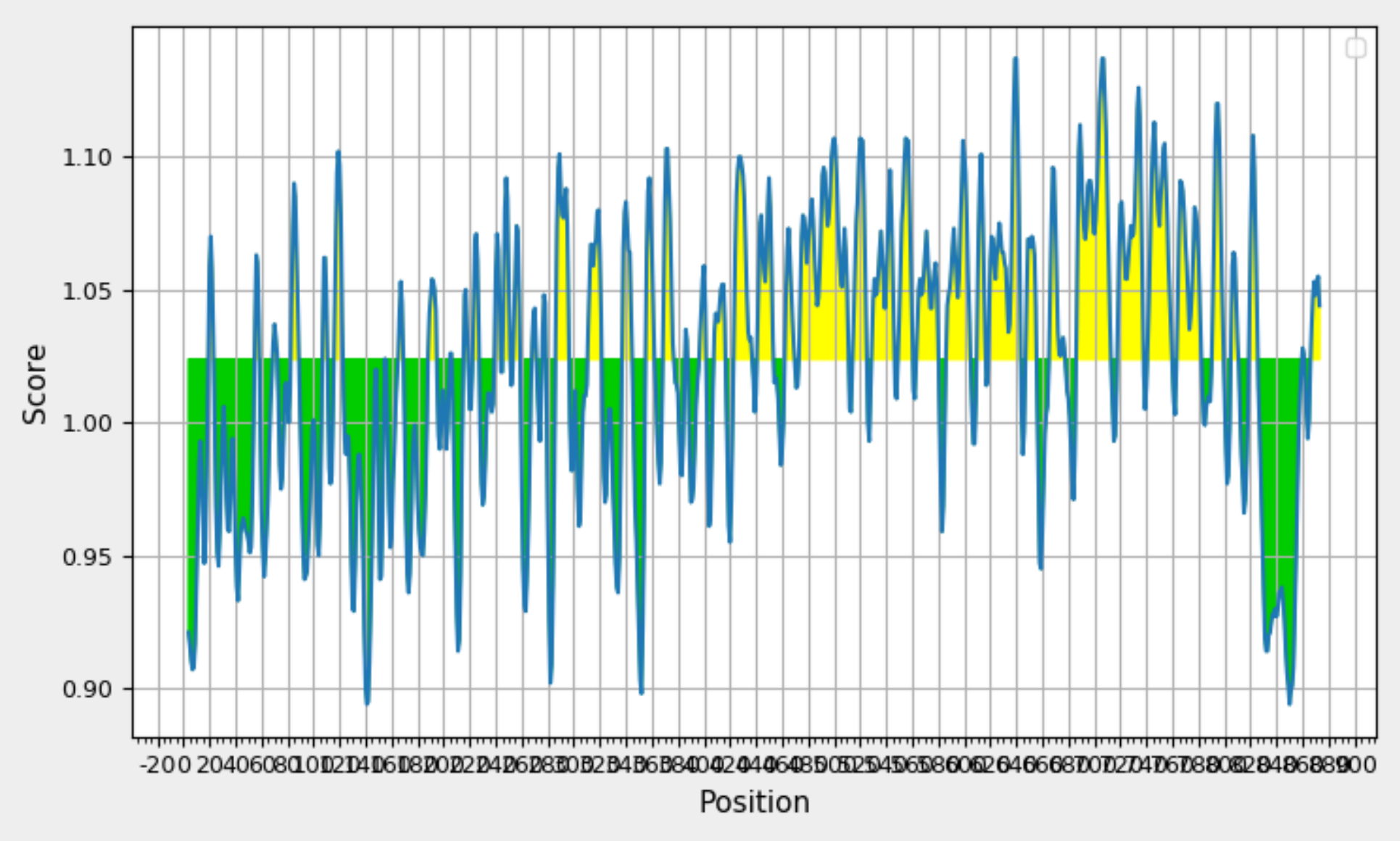
Validation of Antigenicity: Kolaskar and Tongaonkar Analysis Reveals "T41 Glycoprotein 350/220" with a Recognized Threshold Value of 0.879. Yellow Regions Signify Antigenic Properties.


Analyzing B-Cell Epitope Predictions
The identification of B cell epitopes involves considering specific attributes crucial for effective recognition by B cells, including hydrophilicity, surface accessibility, and the prediction of beta-turn structures. In the analysis of the query protein, various web-based tools within the IEDB were utilized for scanning. Specifically, the Kolaskar and Tongaonkar antigenicity prediction tool was employed to assess the physico-chemical properties of amino acids and their prevalence in known B cell epitopes. The results, illustrated in Figure 4 and summarized in Table 4 indicated an average antigenic propensity value of 1.015 for the protein, with a maximum value of 1.220 and a minimum of 0.879. The tool was configured with a threshold value of 1.015 to identify antigenically potent regions, leading to the identification of a 16-mer epitope spanning amino acid positions 260 to 340. This specific region demonstrated favourable characteristics for a B cell epitope, making it a promising candidate for further exploration in the context of vaccine development.
Table 4
Antigenicity Analysis Utilizing the Kolaskar and Tongaonkar Methodology
The critical factor influencing B cell immune response is the surface accessibility of B cell epitopes. Hydrophilic regions exposed on the surface are more likely to trigger a B cell response. In this study, we employed the Emini surface accessibility prediction and Parker hydrophilicity prediction tools to assess surface accessibility. Table 4 presents the results, offering valuable insights into the characteristics of potential B cell epitopes. Additionally, Figure 6; Figure 5 provide visual representations generated by both tools, offering a graphical overview of surface accessibility features identified in the analysed protein sequence. These analyses contribute to a comprehensive evaluation of B cell epitopes, facilitating the selection of regions with optimal surface accessibility for further consideration in vaccine development, as outlined in Table 5 .
Table 5
An "Emini Surface Accessibility Analysis: Unveiling Antigenic Characteristics
Figure 7
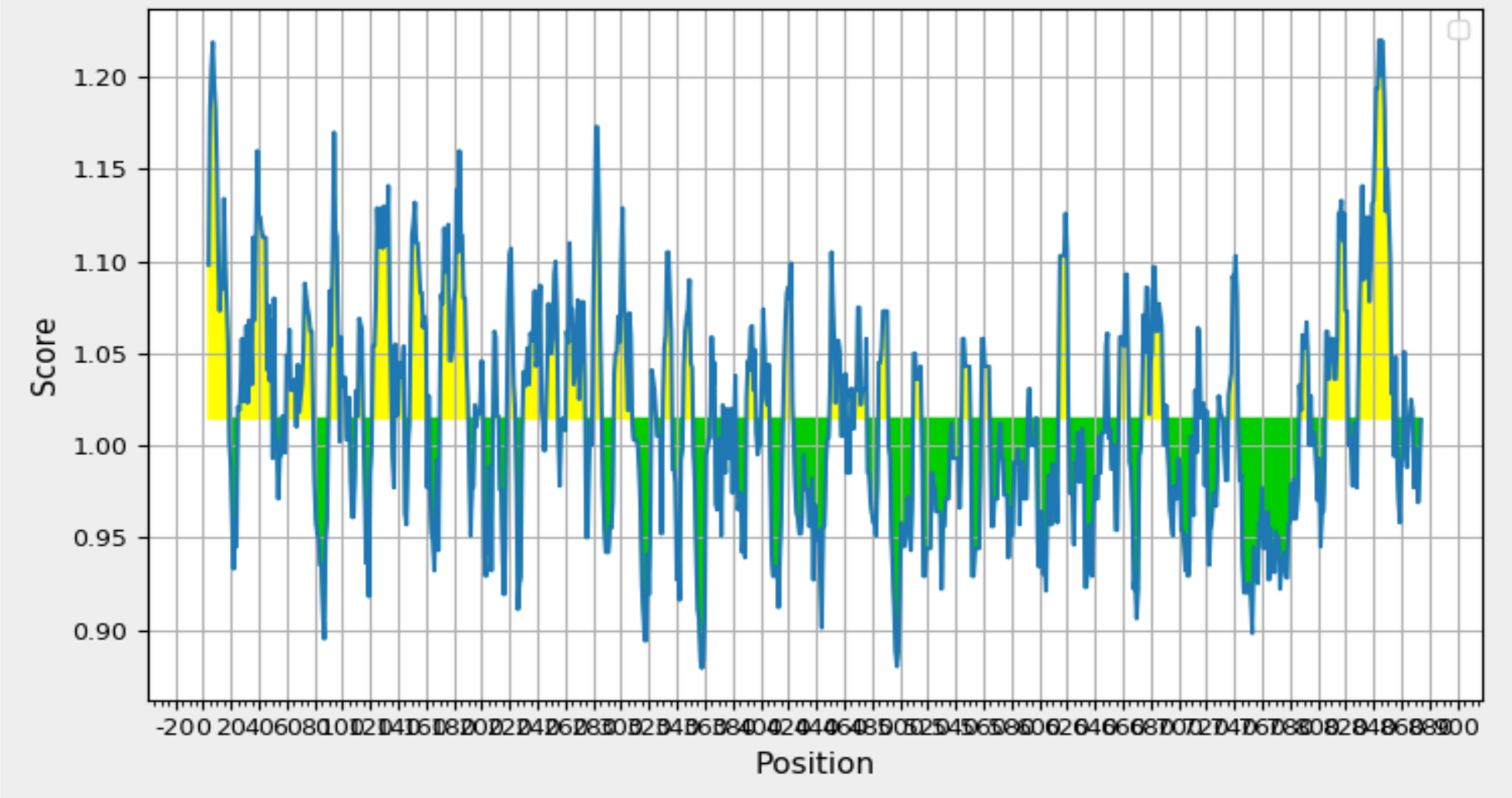
Validation of Elevated Antigenicity: Chou and Fasman Beta-Turn Prediction Highlights Glycoprotein 350/220. Graph Depicting Sequence Position (x-axis) and Surface Probability (y-axis) Identifies Beta Turns in Yellow, Exceeding the 1.130 Threshold.
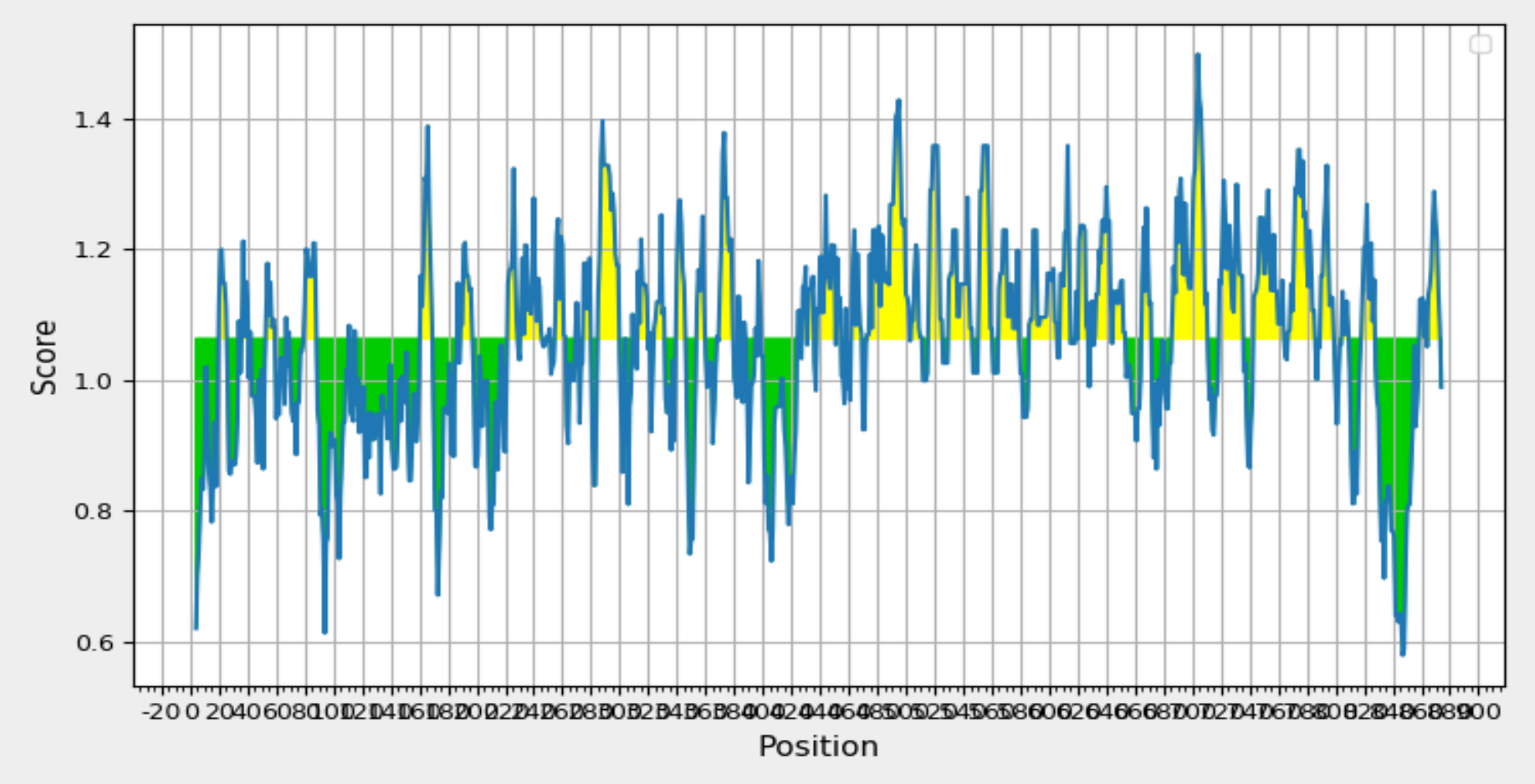
Figure 8
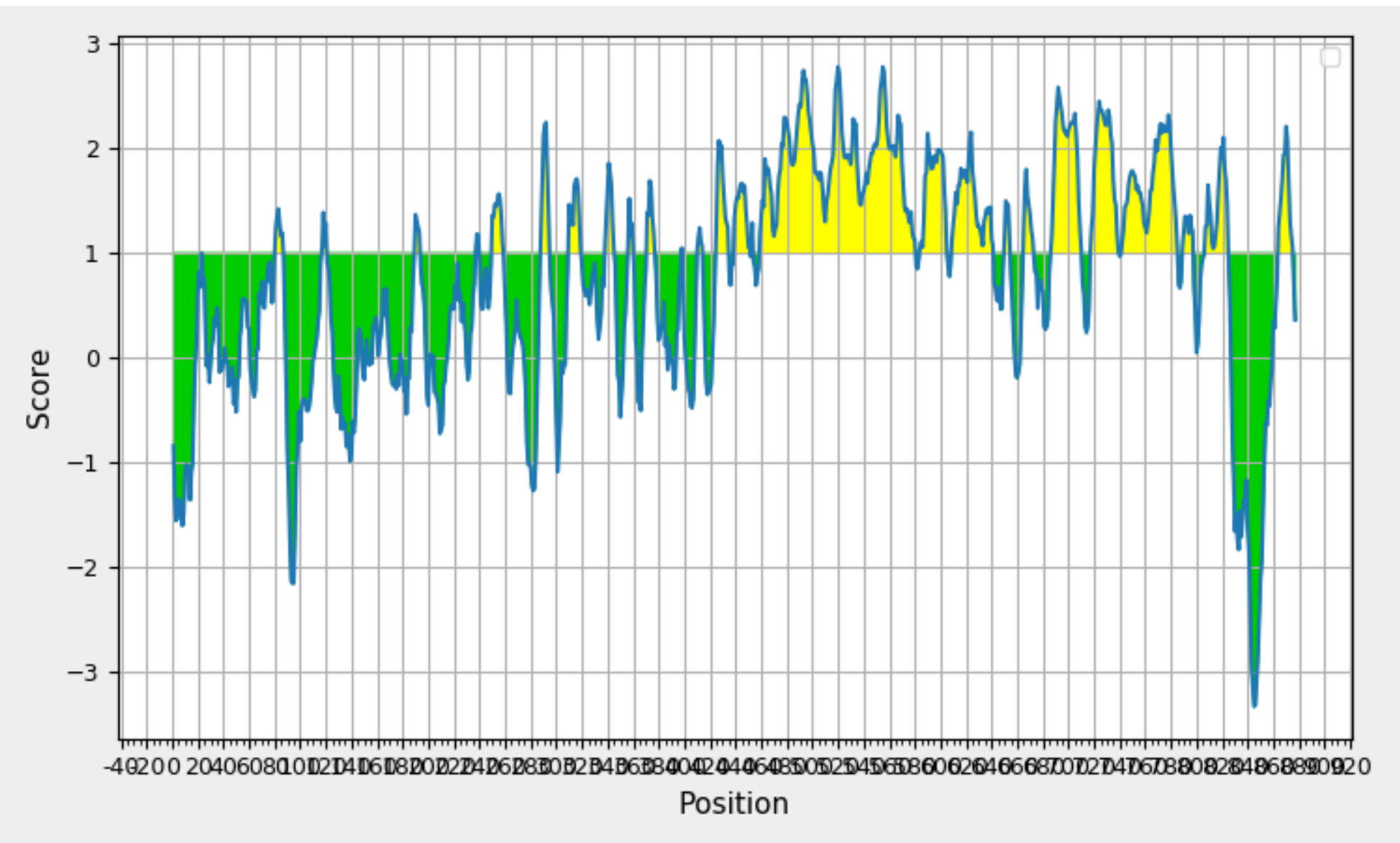
Analyzing Glycoprotein 350/220 Antigenicity:Kolaskar and Tongaonkar Prediction. Graph Depicting Sequence Position (x-axis)and Antigenic Propensity (y-axis) Displays Yellow Highlights Signifying Antigenic Regions Above the 0.98 Threshold.
To identify beta turn regions within the query protein, a Chou and Fasman Beta turn prediction was conducted. The results of this analysis revealed a consistent predicted beta turn region spanning from position 260 to 353 within the protein sequence. Beta turns are known to exhibit characteristics of surface accessibility and hydrophilicity and play a substantial role in inducing antigenicity, making this discovery particularly relevant. Additionally, the analysis of flexibility within the protein was carried out using the Karplus Schulz flexibility prediction tool. The results identified the region spanning from position 342 to 353 as the most favourable in terms of flexibility. Experimental data suggests that the portion of a peptide interacting with an antibody often exhibits a degree of flexibility, adding significance to this finding. Visual representations of these results are available in Figure 8; Figure 7, providing a graphical overview of the predicted beta turn region and the flexible regions within the protein. These findings contribute valuable insights into potential B cell epitopes, aiding in the selection of regions with optimal characteristics for further consideration in vaccine development.
Figure 9
Exploring BepipredLinear Epitope Prediction for the Protein with Maximum Antigenicity. The GraphDepicts Position (x-axis) and Prediction Score (y-axis), with Yellow Shading Signifying Beta Turn-Rich Areas Above the 0.350 Threshold. The Peak Represents the Most Potent B-Cell Epitope.
Bepipred, employing a Hidden Markov model as part of its machine learning process, serves as a valuable tool for the identification of Linear B cell epitopes. The rationale behind its use lies in addressing the limitations of single-scale amino acid propensity profiles, which may not consistently and accurately predict antigenic epitopes. By leveraging Bepipred, researchers aim to achieve improved results in epitope prediction, surpassing the performance of receiver operating characteristic (ROC) plots. The Bepipred-predicted epitopes for the protein are detailed in Table 6, providing a comprehensive overview of the identified epitope regions. A visual representation of these epitopes can be found in Figure 9 , offering a graphical depiction of the predicted B cell epitopes within the protein sequence. Following a thorough analysis of data obtained from various B cell epitope prediction tools, it has been determined that the region spanning from amino acids 347 to 361 is identified as the most promising region for eliciting a B cell immune response. This region is considered particularly favorable for further exploration in the context of vaccine development.
Figure 10
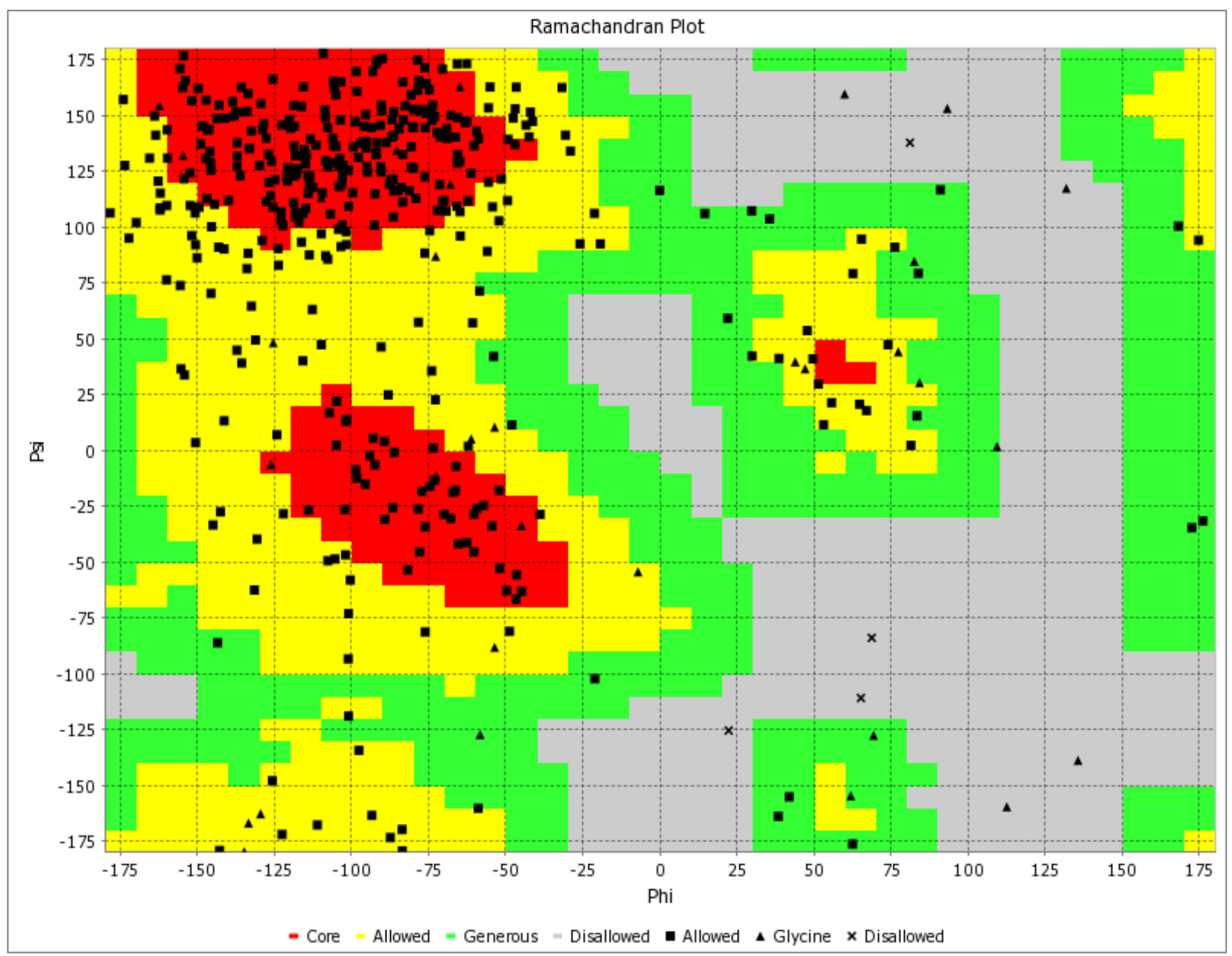
Visualizing the Predicted Model of Glycoprotein 350/220 from Epstein-Barr Virus through a Ramachandran Plot. Distinct Colors Indicate Structural Conformations: Red for Preferred, Yellow for Allowed, Light Yellow for Generously Allowed, and White for Disallowed. Torsion Angles Phi (Φ) and Psi (Ψ) Play a Central Role in Defining the Protein's Structural Orientation.
Molecular Interaction Modeling
The binding free energy assessment of the 'ATNLFLLEL' epitope was conducted using the MM/PBSA method. The MM-PBSA calculations for the protein-ligand complex were performed based on the final 10-ns trajectories using the g_mmpbsa tool integrated into the GROMACS software. Snapshots of the HLA-DRB1*1501 - 'ATNLFLLEL' complex were extracted from molecular dynamics (MD) simulation trajectories between 6000 ps and 8000 ps at intervals of 15 ps, as well as between 4000 ps and 6000 ps, for MM/PBSA calculations. The analysis of these snapshots revealed that only the 'ATNLFLLEL' epitope effectively occupied the receptor and exhibited the lowest binding energy of -3.6 kcal/mol. A smaller binding energy indicates a stronger binding affinity, suggesting the epitope's suitability for vaccine development. To further validate the quality and reliability of the 'ATNLFLLEL' epitope, Ramachandran plot analysis was employed, utilizing the PDB sum generates tool. The refined model demonstrated that the majority of the protein's residues (>90%) occupy the most favored regions of the Ramachandran plot, indicating improved structural quality (Figure 10). Therefore, based on the binding energy assessment and structural analysis, the 'ATNLFLLEL' epitope is considered an efficient candidate for vaccine development.

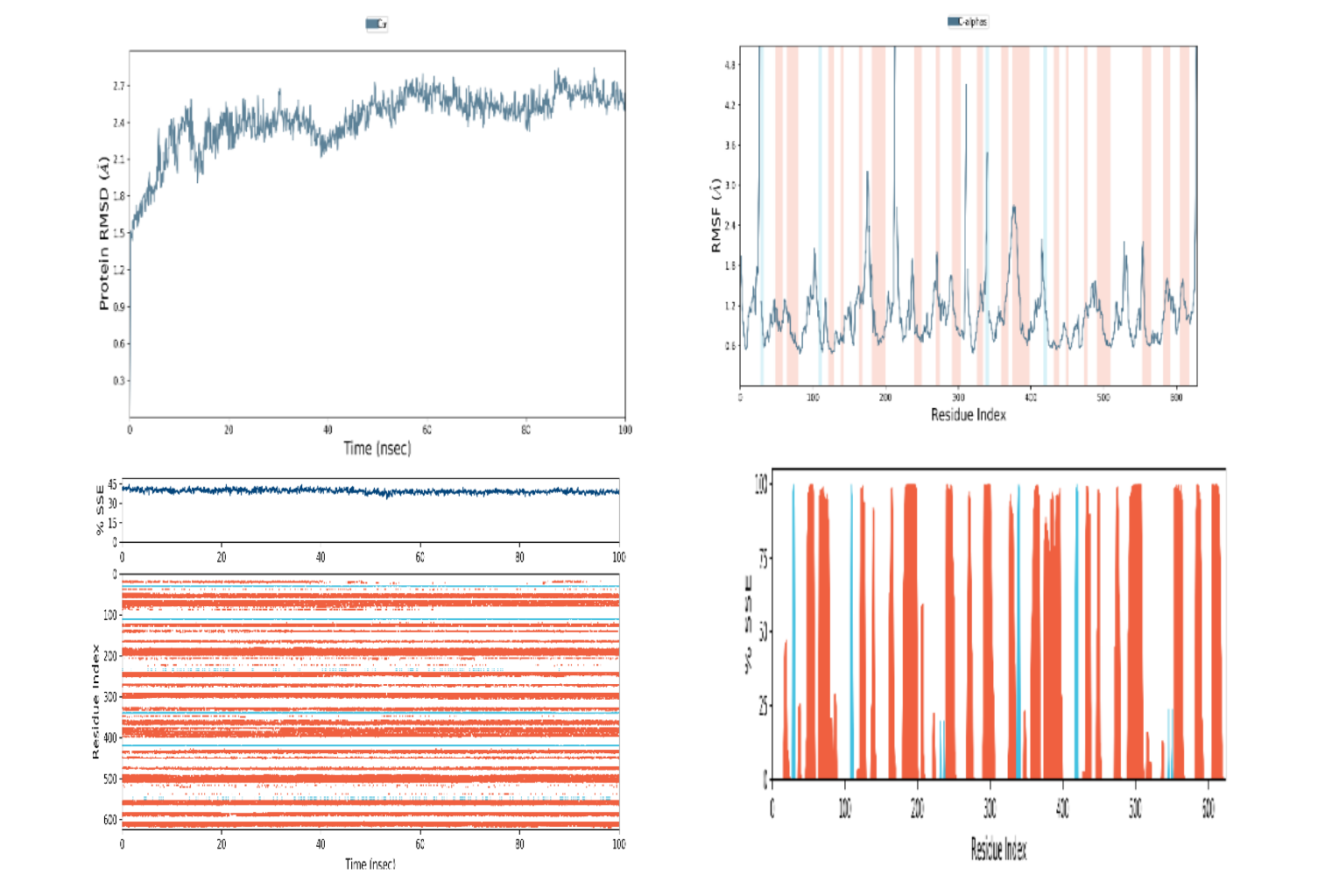
Figure 12
Illustrates the results of molecular dynamic simulations for the developed vaccine. The representation includes: (A) Root Mean Square Deviation (RMSD) plot for Protein-Ligand (PL). (B) RMSD plot for Protein (P). (C) Histogram depicting the Secondary Structure Elements (SSE) of Protein (P-SSE). (D) Timeline representation of the Secondary Structure Elements (SSE) for Protein (P-SSE-Timeline)
Refinement and Construction of the Model
The three-dimensional structure of the Glycoprotein 350/220 from the Epstein-Barr virus was constructed using the Phyre2 server. In molecular biology, understanding a protein's tertiary structure is essential for unveiling its molecular functions and interactions. It's important to note that homology modeling tools, such as Phyre2, may introduce local distortions into the generated models. To address this concern, the model generated by MODELLER underwent additional refinement through ModRefiner, aiming to achieve a more precise stereochemical representation. Subsequent analyses were conducted utilizing this refined model. For reference, Figure 11 presents a visualization of the 3D model generated by Swiss-PDB. This refined three-dimensional model provides a basis for further investigations into the structural and functional aspects of the Glycoprotein 350/220, aiding in a more comprehensive understanding of its role in the Epstein-Barr virus and potential applications in vaccine development or therapeutic interventions.
In the course of this investigation, a molecular dynamics simulation was executed to assess the stability of the predicted epitope 'ATNLFLLEL' during the formation of a protein complex. The GROMACS 5.0 package was utilized for this purpose, allowing for the evaluation of several crucial structural metrics. Key parameters included the Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Residual Index (Ri). The computed RMSD value, which represents the overall deviation of the complex from its reference structure, was measured at 0.49 nm. Simultaneously, the RMSF score, indicating local structural fluctuations within the complex, was determined to be 0.12 nm. These results collectively suggest a high level of stability within the formed complex, indicating that the predicted epitope 'ATNLFLLEL' forms a robust interaction with the associated protein (Figure 12). The stability observed during the molecular dynamic’s simulation enhances the confidence in the reliability of the predicted epitope for further consideration in vaccine development or related applications.
Discussion
In the current world, illnesses stand as a widespread global challenge. With the growing emergence of new viruses affecting people worldwide, the rapid development of vaccines has become exceptionally important to effectively reduce the increasing incidence of viral assaults (Graham & Sullivan, 2018). In recent times, the emergence of advanced sequencing technologies and the progress in genomics and proteomics have caused a noteworthy transformation in the realm of computational immunology." Thanks to an unparalleled wealth of accessible information, modern immunoinformatics tools are rapidly advancing in both development and deployment. These instruments enable a forward-thinking stance in vaccine creation, harnessing an enriched understanding of the immune responses of the human body to a diverse spectrum of organisms. Persistent efforts to counteract the aggressive actions of Epstein-Barr Virus have taken various forms, involving a range of therapeutic strategies (Andrei, Trompet, & Snoeck, 2019). These approaches extend from crafting small molecule inhibitors to formulating vaccines. Antagonists primarily focus on critical viral proteins, while vaccines seek to trigger immune responses from B-cells or T-cells against both lytic and latent EBV proteins (Silva, Alves, & Pontes, 2024). Despite the challenges in finding definitive cures for EBV-related infections, considerable headway has been achieved in clinical trials of a gp350 vaccine. It's crucial to underscore that while this vaccine demonstrates notable progress, it doesn't entirely prevent viral infections. Scientific inquiry has unveiled EBV's significant role in the development of certain tumors (Su, Siak, Leong, & Cheah, 2023). EBV infections have been implicated in various human diseases, including cancer and autoimmune disorders (Houen & Trier, 2021). The specific reasons why some individuals, despite displaying appropriate responses to EBV, develop associated diseases while others don't, remain elusive. Nonetheless, it is clear that genetic and environmental factors, encompassing lifestyle choices and prior encounters with pathogens, contribute to this variability. Given the absence of a pharmacological cure for EBV and the absence of available preventive or therapeutic vaccinations, the prospect of developing a prophylactic EBV vaccine holds promising potential in averting EBV-associated diseases (Cai et al., 2021). The EPV virus, with a diameter ranging from 122 to 180 nm, consists of a double helix of deoxyribonucleic acid (DNA) containing approximately 172,000 base pairs encoding 85 genes. This DNA is enclosed within a protein nucleocapsid, which is further enveloped by a tegument composed of proteins (He et al., 2020). The entire structure is enveloped in a membrane that includes lipids and surface projections of glycoproteins, crucial for infecting the host cell. A promising approach to prevent, delay, or alter the progression of various EBV-associated diseases is vaccination. The focus of developing an anti-EBV vaccine center on the MA complex of EBV. This complex, found on both the envelopes of viral particles and the surfaces of EBV producer cells, consists of a major envelope glycoprotein known as gp350/300 (gp350) and a related glycoprotein called gp250/200 (gp250) (Kanekiyo et al., 2015). The molecular weights of these glycoproteins may vary slightly among different laboratories. Both glycoproteins are products of the same gene, mapped to the BamHI L DNA fragment of the B95-8 genome (Biggin, Farrell, & Barrell, 1984).. The mRNA of gp250 is generated by splicing an internal portion of the mRNA of gp350, maintaining the same open reading frame. gp350 plays a role in virion binding to human B cells through its specific interaction with the receptor for the complement C3d fragment (Khyatti, Patel, Stefanescu, & Menezes, 1991). Upon natural infection with EBV, the majority of individuals develop a neutralizing antibody response, although the titers are generally moderate. The effectiveness of vaccine-induced immunity often hinges on the quality of the immune response, regardless of its magnitude. Notably, the soluble gp350 ectodomain and D123 exhibit limited efficacy in inducing neutralizing antibodies in mice, in contrast to the D123 nanoparticles, despite similar antigenic profiles in both soluble and nanoparticle forms of gp350 (Sun, Chen, Kang, & Zeng, 2021). This underscores the significance of antigen presentation, specifically the conformational authenticity of functional domains, in shaping the immune response's quality. The immune focusing achieved through nanoparticle presentation of specific gp350 domains enables the generation of antibodies predominantly targeting CR2BS, offering a mechanism for potent neutralization in sera (Kanekiyo et al., 2015). Similar modes of neutralization have been described in other viruses, such as the VRC01-class antibodies neutralizing diverse strains of HIV-1 by binding to the CD4-binding site and certain monoclonal antibodies neutralizing viruses like influenza A, hepatitis C, poliovirus, herpes simplex virus, and SARS coronavirus by targeting conserved sites (Li et al., 2011). Given the functional conservation required for the receptor-binding site (RBS) to engage its host cell receptor, it emerges as one of the virus's most vulnerable targets (Schmidt et al., 2015). The initial findings in anti-EBV vaccine studies are promising; however, there is a need for enhancements in terms of immunogenicity and the selection of vaccine epitopes to strive for a more effective vaccine. Consequently, new vaccine formulations have been devised. Regarding prophylactic vaccines, gp350/220 was the first identified and remains the most extensively researched vaccine target. Yet, glycoproteins gH/gL, gp42, and gB have also been recognized as targets for neutralizing antibodies (Jean-Pierre, Lupo, Buisson, Morand, & Germi, 2021). Moreover, there is ongoing exploration of mixed approaches that involve both EBV lytic cycle and viral cycle proteins in vaccine development. Consequently, there has been a discernible surge in research initiatives that are mostly focused on developing a vaccine against the EPV virus (Zhong et al., 2022). Nevertheless, the results to date in this field are still unclear. The goal of this work is to use immunoinformatics technologies to discover critical immunogenic epitopes on Powassan virus proteins. Predicting the composition of a prospective vaccination that targets the T41 Glycoprotein 350/220 is the aim. Because entire pathogens were used in live, attenuated, or inactivated forms in earlier immunization techniques, there was a risk of pathogen reactivation. Regional genetic variability may lessen the effectiveness of vaccinations. Innovative strategies that minimize complexity and adverse effects while eliciting targeted immune responses, such as DNA vaccines and epitope-based vaccinations, provide answers. DNA vaccines target the immune system without infecting the recipient by exploiting the genetic material of the disease. Vaccines based on epitopes target immunogenic areas of pathogens to maximize immune responses against vital components. These developments enhance the efficacy and safety of vaccines, which is essential for fighting viral illnesses. Using in silico methods to build neutralizing peptide-based vaccines has been efficacious against a range of viruses. T-cell epitope-based vaccines seek to elicit strong CD8+ T-cell mediated immunological responses, whereas the majority of epitope-based vaccinations primarily stimulate B-cell epitopes. By addressing the constraints of antigenic drift, T-cell-based vaccinations, including multi-epitope vaccines incorporating B and T-cell epitopes, provide intriguing options. With high conservation (83.66%) among sequenced EPV T41 Glycoprotein 350/220 proteins, the chosen T-cell epitope, "ATNLFLLEL," may be a contender for wider protection. Its compatibility with a variety of HLA profiles is improved by its interaction with eight HLA-A and HLA-B alleles. An examination of population coverage reveals encouraging responsiveness across a range of geographies, highlighting its potential as a vaccine component with broad applicability. The epitope's non-allergic classification also lends credence to its possible efficacy for a wide range of individuals. Its significant affinity for MHC-I molecules is confirmed by docking studies with HLA-B*3501, so establishing its status as a new vaccination candidate. Analysis revealed an area from amino acid positions 253 to 310 that contains possible B-cell epitopes. Of these, the sequence "LGAGELALTMRSKKLP" has been identified as a promising epitope because of its antigenic potential. Pre-therapy vaccine development for in vitro and in vivo investigations is emphasized in the study, given the chronic nature of HPV infection and the need for therapeutic options. With potential T-cell and B-cell epitopes found for a multipeptide vaccination, EPV T41 Glycoprotein 350/220 shows promise as a target for therapeutic development.
This computational study underscores the importance of extensive in vitro validation and experimentation, establishing a groundwork for future research endeavors. While the findings offer insights into the development of potential drugs and vaccines against HPV and EPV, it is crucial to emphasize the necessity for further laboratory-based methods to validate these outcomes.
Author contributions
Dr. Saba Beigh conceived the structure of this research article, investigated key resources for the research, edited, and revised the manuscript. Dr Saba Beigh revised, read and agreed to the published version of the manuscript.
Abbreviations
EBV: Epstein–Barr virus; gp350: glycoprotein gp350 ; HLA: Human leukocyte antigens; MHC: major histocompatibility complex ; AS04: Adjuvant system 04; CR2: complement receptor type 2; BGLF5:Epstein–Barr Virus (EBV) DNase; BZLF1: BamHI Z Epstein-Barr virus replication activator; (CR2/CD21): complement receptor 2; CTLs: cytotoxic T lymphocytes; HIV: human immunodeficiency virus; CD4+/CD8+ ratio: T helper cells (with the surface marker CD4) to cytotoxic T cells (with the surface marker CD8); NCBI: National Center for Biotechnology Information; IEDB: The Immune Epitope Database; WHO: World Health Organization.