

Introduction
Opioids are a class of narcotic drugs that induce sleep and act as analgesics in the human body. As much as the positive impact it offers to mankind, its addictive nature may cause more harm and can even be fatal in extreme cases (Vuong et al., 2010). Drug addiction or substance abuse presents a perplexing challenge that confronts every society worldwide. As per the United Nations Office on Drugs and Crime Estimate, 6 million people in the age group between 15 and 64 years are using opioids globally to stimulate ecstasy. The number of opiate users are 3.15 million on a similar ground (Nations, 2023). Opioids are often prescribed as analgesics for pain management (Dickenson, 1991). In addition to its therapeutic potency, opiates give a euphoric feeling (Mcauliffe, Rohman, Feldman, & Launer, 1985). Opiate abuse has been associated with persistent cognitive impairments in humans. While some research has indicated that these declines in cognitive function might be connected to impaired frontal lobe activity, other studies focusing on the impact of opiate exposure on the structure of the hippocampus and its role in memory and learning suggest that changes in hippocampal function may also contribute to the cognitive difficulties observed in individuals who abuse opiates (Kutlu & Gould, 2016).
In 2009, Letizia Paoli et al. said in their paper that India could be the third largest illicit opium-producing country after Afghanistan and Burma based on their research (Paoli, Greenfield, Charles, & Reuter, 2009). In 2021, opioid overdoses claimed the lives of more than 80,000 people, with synthetic opioids being responsible for nearly 88% of these tragic deaths. Opium and related drugs originating in Afghanistan are illicitly transported into several Indian states, including Gujarat, Rajasthan, Punjab, and Jammu Kashmir (CDC, 2021). The trafficking of opiates from the Golden Triangle (Myanmar, Thailand, and Laos) region has decreased in recent years. This decline can be attributed to the local cultivation of poppy plants in various Indian states such as Himachal Pradesh, Manipur, Jharkhand, Arunachal Pradesh, and Uttar Pradesh (Patil & Pandey, 2022).
The receptors that bind the opioids are the mu receptor (MOR), kappa receptor (KOR), delta receptor (DOR), and nociception receptor (NOR). The most frequently employed opioids for pain management primarily affect the μ opioid receptor (MOR) systems. Although MOR remains highly effective as an analgesic, it also possesses the ability to enhance mood and trigger the activation of central dopamine reward pathways, leading to the modulation of euphoria. These undesirable side effects have motivated researchers at both basic and clinical levels to actively explore other opioid receptors as potential targets for developing pain relief medications. Traditional signaling pathways of opioid receptors involve their presence in various areas of the brain, including the medulla, locus coeruleus, and periaqueductal gray region, which are involved in pain modulation (Al-Hasani & Bruchas, 2011). These receptors are also found in limbic, midbrain, and cortical structures (Al-Hasani & Bruchas, 2011). When these opioid receptors are activated in these locations, they directly hinder neurons, leading to the inhibition of pain signals transmitted through the spinal cord (Al-Hasani & Bruchas, 2011).
Molecular Docking is the common method of drug discovery. Molecular docking came into light in the year of 1980’s. It utilizes different algorithms to perform molecular docking studies. It is an essential tool in the field of pharmaceutical research (Meng, Zhang, Mezei, & Cui, 2011). It is mainly used to model a protein-ligand interaction at the atomic level. This helps in characterizing the behavior of the ligands in the target protein’s binding site. The two basic steps of molecular docking are prediction of ligand-protein conformation after the binding and its orientation; and the assessment of binding affinities (Meng et al., 2011).
Molecular docking mainly helps in drug discovery but it may help forensic scientists in many aspects (Govindaiah, Sreenivasa, Ramakrishna, Rao, & Nagabhushana, 2018). Molecular docking plays a pivotal role in forensic science by facilitating the elucidation of complex molecular interactions between forensic evidence and potential suspects or relevant substances. This computational technique is indispensable for forensic investigations due to its ability to predict the binding affinity and interaction patterns between small molecules, such as drugs, toxins, or metabolites, and their respective target biomolecules, often proteins or nucleic acids.
Molecular docking mainly performs two tasks: i) sampling ligands in the active site of protein by scoring methods ii) and ranking the conformations by the scoring method. The scoring method rather than calculating, estimates the conformations of the complexes (Meng et al., 2011).
There are three docking methodologies performed: i) Rigid Ligand and Rigid Receptor Docking- in this case, the ligand and the receptor are considered as rigid bodies which means the search space might be limited. Rigid in terms of simulation implies that the bodies may have fixed shapes throughout the process (Meng et al., 2011). ii) Flexible Ligand and Rigid Receptor Docking- here, the ligands are set to be flexible, meaning, the docking process considers the fact that, the complexes may change their shape according to how they are bound to each other. AUTODOCK and many other software have adopted this semi-flexible docking method (Meng et al., 2011). iii) Flexible Ligand and Flexible Receptor Docking- In this, both the ligand and receptor are set as flexible (Meng et al., 2011).
In forensic toxicology, molecular docking enables the precise modeling of ligand-receptor interactions, allowing forensic scientists to assess the plausibility of various scenarios involving toxic substances. By virtually docking candidate compounds with specific cellular targets, forensic experts can infer toxicological mechanisms, estimate the likelihood of intoxication, and discern the chemical basis behind observed physiological effects. This paper is going to utilize molecular docking tools to predict the mu-opioid receptor orientation to different opiates and opioids. The complex with the lowest binding energy is considered to be in the best conformation.
Materials and Methods
Software and Resources
To perform the docking, the appropriate software was downloaded from the internet. The required softwares were Python version-3.11.5, MGL Tools version-1.5.7 (Sanner, 1999), AutoDockTools version-1.5.7 (Sanner, 1999) and Babel version-2.4.1 (Boyle et al., 2011). Python is a high-level programming language that makes the developers easy to understand the code. MGL Tools, also known as MGLTools, is a software package used in the field of computational molecular biology and chemistry. It is designed to assist researchers, scientists, and professionals in the study of molecular structures and interactions, particularly in the context of drug discovery and molecular modeling. AutoDock is a suite of molecular docking tools used in the field of computational chemistry and molecular biology. The results were analyzed in a software called BIOVA Discovery Studio Visualizer v21.1.0.20298 (Biovia, Systèmes, & Workbook, 2023). The ADME analysis was done by the SwissADME tool (Daina, Michielin, & Zoete, 2017).
Blind Docking
The concept of blind docking was first introduced by Hete ́nyi and Spoel in 2002 with the help of the Autodock program (Hetényi & Spoel, 2002).
Protein Selection and Preparation:
Firstly, the mu-receptor protein of Homo sapiens was downloaded from the RCSB PCB website. The downloaded receptor protein is 6DDF (Koehl, Hu, & Maeda, 2018), Mu Opioid Receptor-Gi Protein Complex. There were many other MOR protein structures available but 6DDF was chosen because of the less complexity of its structure. The other MOR protein structures had more agonists and antagonists ligands bound to it already which made the structure more complex. To understand the best binding sites of a receptor protein for conducting the docking, it is essential to perform a blind docking.
Ligand Preparation:
As a ligand, the narcotic drug morphine was taken for doing the blind docking. The morphine structure was downloaded from the PubChem database in sdf-MDL MOL format. Since Autodock Tool 1.5.7 doesn’t read this format, the format of the morphine was converted from sdf- MDL MOL format to PDB format and saved the file. Then the blind docking process was started by opening the Autodock Tools 1.5.7 software.
Docking simulation
The blind docking was done in this research primarily to know the active sites of the MOR protein receptor, which would enable us to perform the site-specific docking. However, there are previous studies that have performed blind docking on MOR receptors, namely, PDB ID 4DKL but they belonged to non-human organisms; as this study mainly focuses on the binding of opioids to the human bodies, blind docking was performed. Moreover, the selected protein is Gi protein complex which is a membrane protein that enables the activation or inhibition of the appropriate mechanisms of the ligands bound to it.
In that, first, the protein structure was to be retrieved. After retrieving the protein structure, water molecules were deleted so that they wouldn't interrupt the docking. The downloaded protein had ligands DAMGO and MEA bound to its structure, they were removed. Then, polar hydrogen molecules were added, followed by the addition of Kollman charges. The file was saved in PDBQT format and the ligand Morphine structure was retrieved. The root in the ligand was detected and the file was saved in PDBQT format. After the retrieval of both structures, a grid box was set with parameters enough to have the protein structure in it and was saved in GPF format. The docking search parameter was set to be the genetic algorithm and the Lamarckian output was saved as a DPF file. Finally, the docking was run. The result as shown in Table, was generated in a Word file with DLG format which gives all the relevant information including, the final genetic algorithm docked state of each conformation, and the 10 best conformations possible for that single protein structure with their binding energies. The conformation with the lowest binding energy was saved in .pdbqt format. As for viewing the complex, a software called Discovery Studio was installed. The saved complex was viewed for the analysis and the 2D structure as shown in Figure 1, was analysed. From the 2D complex the active amino acids were noted and we proceeded to do the site-specific docking of the opiates and synthetic opioids.
Site-Specific Docking
A total of 18 drugs were docked. This study includes five natural and thirteen synthetic opioids as these were the opioids that were mentioned in several research papers (Wang, He, Wang, Wen, & Cao, 2023) and review papers (Pérez-Mañá et al., 2018), (Gutwinski, Schoofs, & Stuke, 2016). The natural opiates that were selected for docking were Codeine, Morphine, Noscapine, Papaverine, and Thebaine. The synthetic opioids docked were Buprenorphine, Carphentanyl, Fentanyl, Hydrocodone, Hydromorphone, Levomethadone, Methadone, Oxycodone, Oxymorphone, Pethidine, Remifentanil, Tapentadol, and Tramadone. Each of the drugs was docked similarly to how the blind docking was done. Each docking was given the parameters of 44 x-points, 32 y-points, and 46 z-points as standard and precise docking. Each DLG file was generated and the best conformation was analyzed from the RMSD tables. The complex with the best conformation was saved and viewed in Discovery Studio software. The parameters of Aromaticity, hydrogen bonds, and charges were analyzed. After the analysis of the complexes, the ligands were analyzed through ADME software, SwissADME.
ADME Analysis
ADME analysis is done to describe the absorption, distribution, metabolism, and excretion of drugs (Domínguez-Villa, Durán-Iturbide, & Ávila-Zárraga, 2021). It is mainly utilized in drug development as M. Abdullahi and S. E. Adeniji in their paper have mentioned ADME analysis is essential in drug discovery as it helps rational decision-making of whether the inhibitors can be administered in the biological system or not (Abdullahi & Adeniji, 2020). ADME analysis further helps to know about the ligand properties and their effect on the human body.
Results and Discussion
Blind Docking
The blind docking of the MOR protein with morphine showed 10 possible conformations as depicted in Table 1. The result showed predicted binding sites with binding energies in the range of -4.96 to -6.75, out of which the 3rd conformation was found to have the maximum binding affinity of -6.75. Hence, the 3rd conformation was selected as the best binding site.
The analysis of 3rd conformation in Autodock software showed the details of the conformation. It showed ligand efficiency of -0.32, inhibition constant of 11.35, intermol energy of -7.34, electrostatic energy of -0.86, total internal of -0.68, torsional energy of 0.6, unbound energy of -0.68 and refRMS of 188.43.
Table 1
Showing the RMSD table for the blind docking of MOR protein with morphine.

Site-specific Docking
Common features
(i) Aromaticity- All the protein-ligand complexes showed high aromaticity. However, some complexes showed extra high aromaticity; they are Papaverine, Buprenorphine, Carfentanyl, Fentanyl, Levomethadone, and Remifentanil.
(ii) Charge- All the complexes were shown to have a neutral charge. And each complex has a negative charge on the left-hand side. And a slight positive charge on the right side of the complex.
(iii) Hydrogen bonds- All the complexes expressed the presence of moderate amount of hydrogen bonds.

Figure 3
Two- dimensional chemical structures of the 18 drugs with MOR obtained from Discovery Studio. (from right) 1) Codeine, 2) Morphine, 3) Noscapine, 4) Papaverine,5) Thebaine, 6) Buprenorphine, 7) Carfentanyl, 8) Fentanyl, 9) Hydrocodone, 10)Hydromorphone, 11) Levomethadone, 12) Methadone, 13) Oxycodone, 14)Oxymorphone, 15) Pethidine, 16) Remifentanil, 17) Tapentadol, 18) Tramadol.
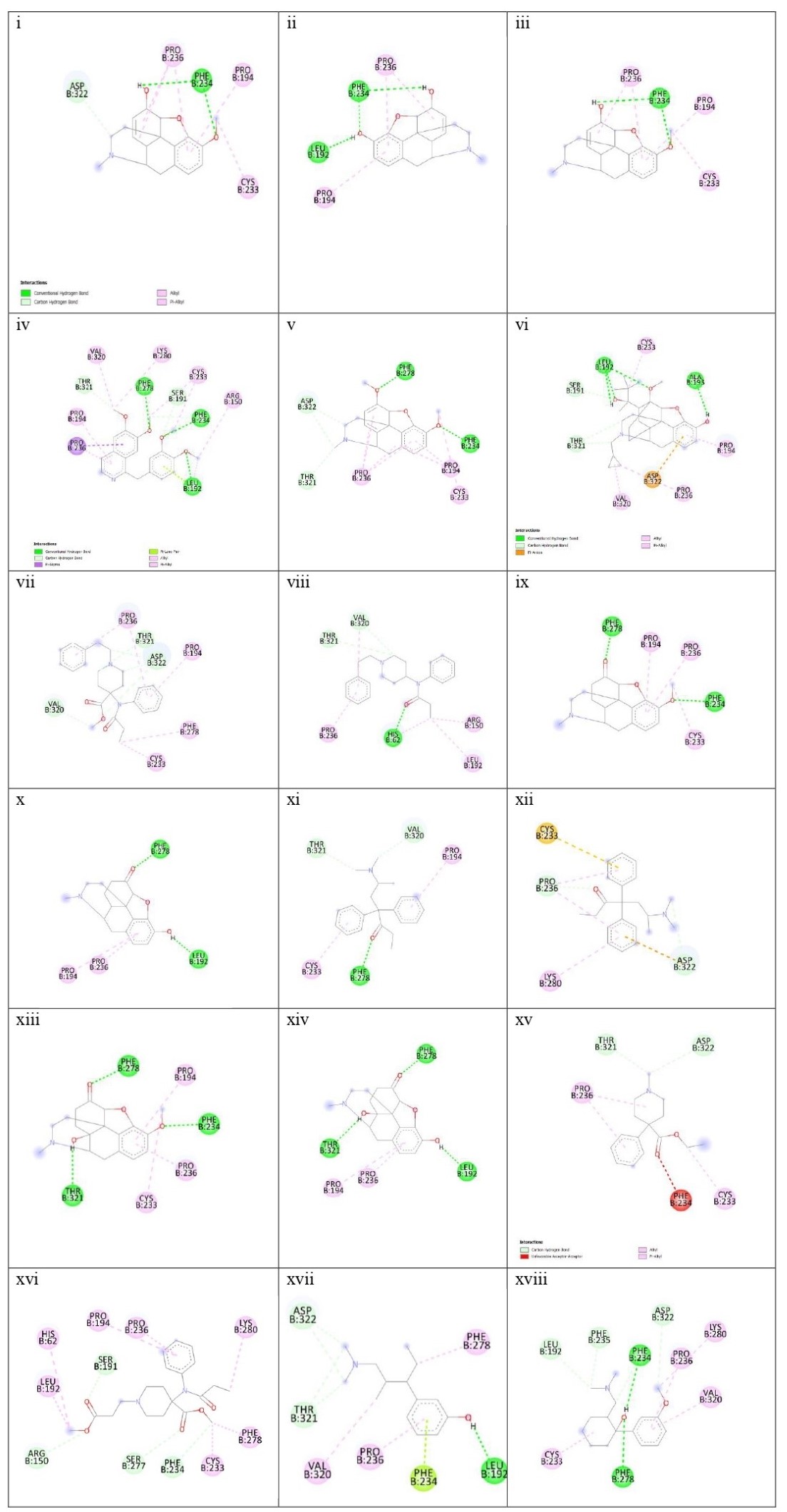
Figure 4
Figure showing aromaticity of the following drugs with MOR. 1) Codeine, 2) Morphine, 3) Noscapine, 4) Papaverine,5) Thebaine, 6) Buprenorphine, 7) Carfentanyl, 8) Fentanyl, 9) Hydrocodone, 10)Hydromorphone, 11) Levomethadone, 12) Methadone, 13) Oxycodone, 14)Oxymorphone, 15) Pethidine, 16) Remifentanil, 17) Tapentadol, 18) Tramadol.
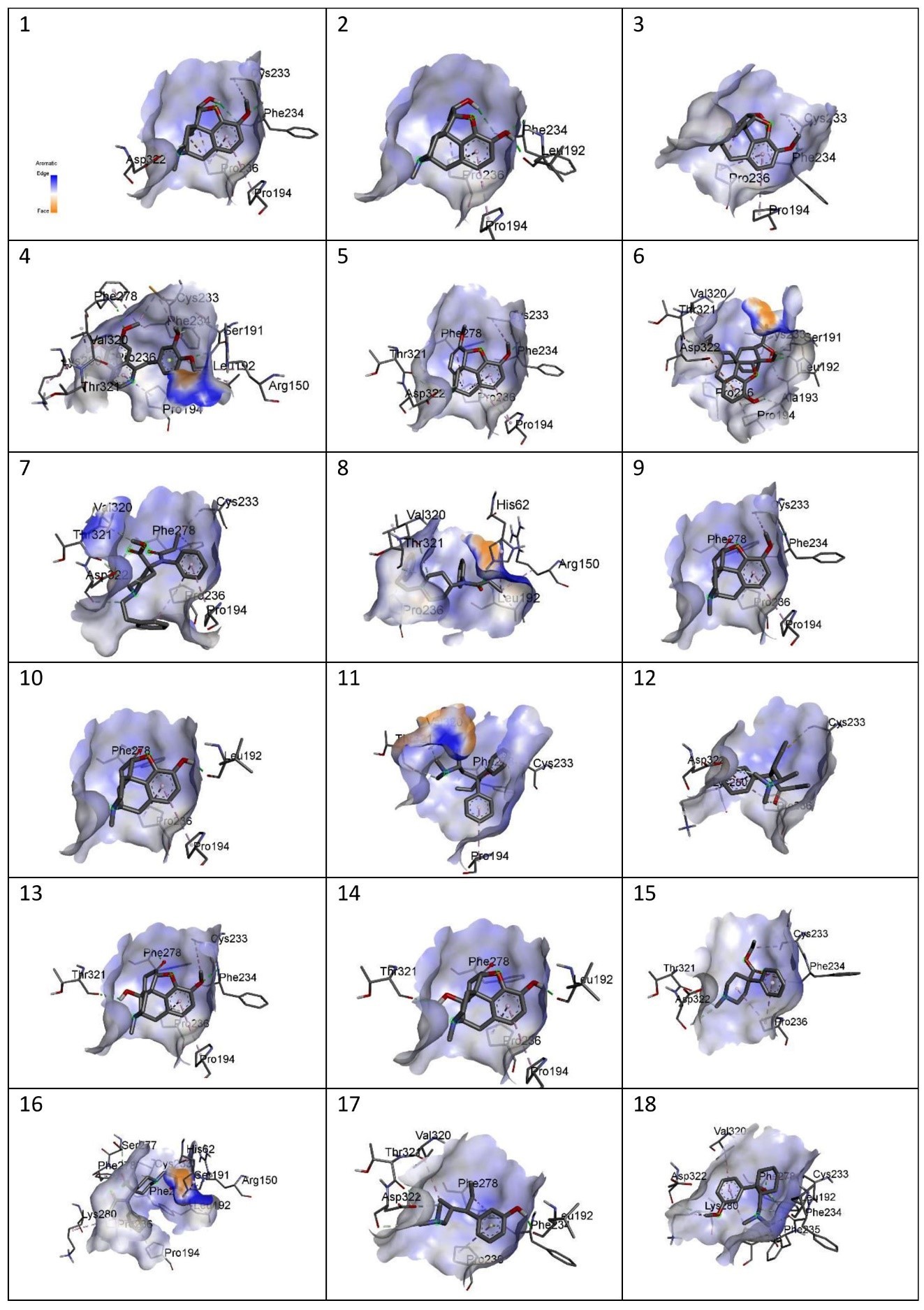
Figure 5
Figure showing charges of the following MOR-ligand complexes: 1) Codeine, 2) Morphine, 3) Noscapine, 4) Papaverine,5) Thebaine, 6) Buprenorphine, 7) Carfentanyl, 8) Fentanyl, 9) Hydrocodone, 10)Hydromorphone, 11) Levomethadone, 12) Methadone, 13) Oxycodone, 14)Oxymorphone, 15) Pethidine, 16) Remifentanil, 17) Tapentadol, 18) Tramadol.
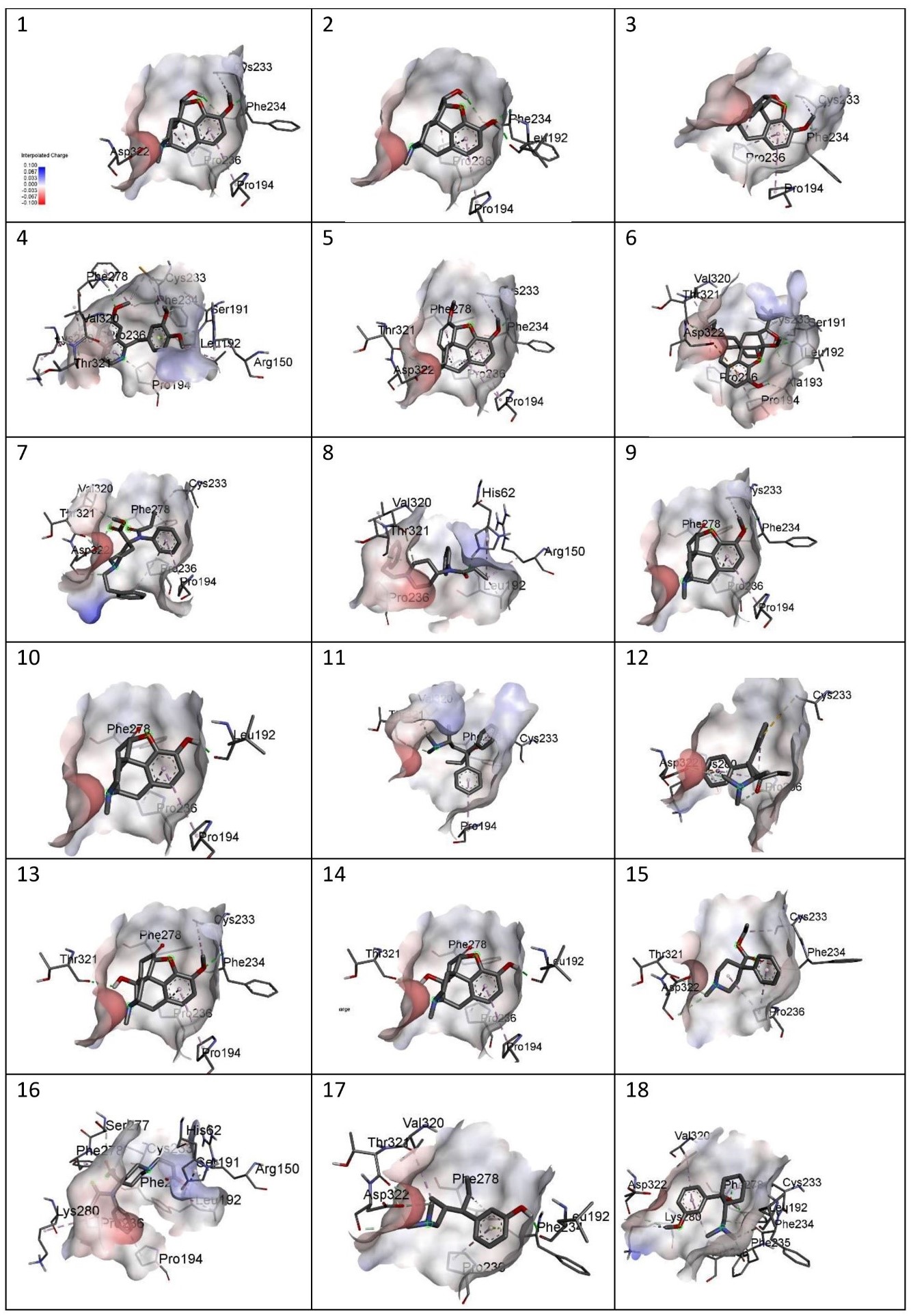
Figure 6
Figure showing the Hydrogen Bonds of the following drugs with MOR: 1) Codeine, 2) Morphine, 3) Noscapine, 4) Papaverine,5) Thebaine, 6) Buprenorphine, 7) Carfentanyl, 8) Fentanyl, 9) Hydrocodone, 10)Hydromorphone, 11) Levomethadone, 12) Methadone, 13) Oxycodone, 14)Oxymorphone, 15) Pethidine, 16) Remifentanil, 17) Tapentadol, 18) Tramadol.
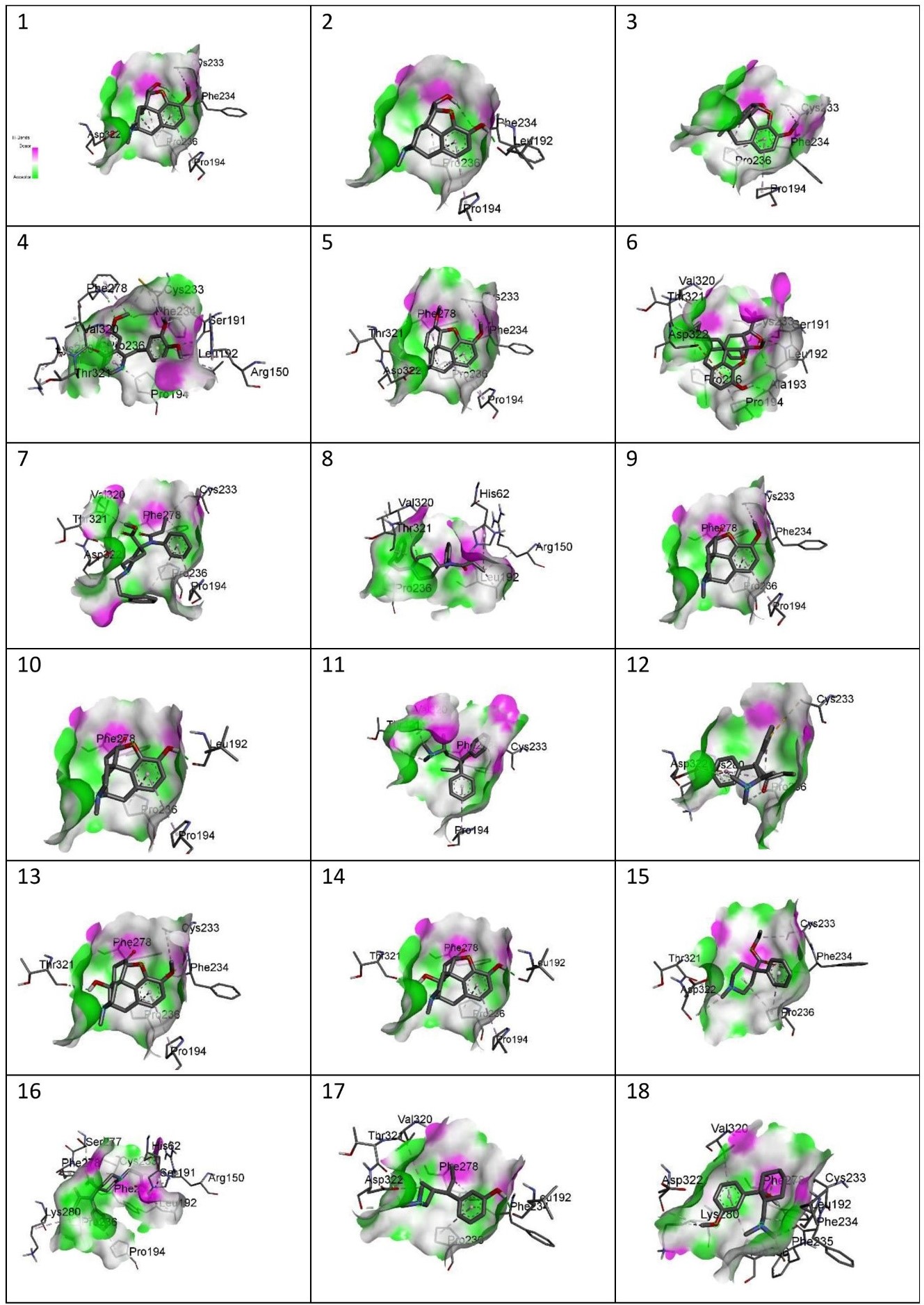
Different features of the complexes
The site-specific docking showed various information about their conformations, as represented in Figure 6; Figure 5; Figure 4; Figure 3; Figure 2. The analysis of the natural opiates is as follows.
Codeine-MOR complex showed its lowest binding energy at -7.82 in its 1st run. Its 2D structure indicated 2 conventional H-bonds with PHE:236 amino acid and 1 carbon H-bond with ASP:322. PRO:236, PRO:194, and CYS:233 were involved with the ligand in alkyl and pi-alkyl bonds. The Discovery Studio image of the complex further revealed a moderate amount of aromaticity present in the complex with a neutral charge. The presence of the aromaticity indicates the non-polar nature of the ligand which further indicates they are hydrophobic. Thus, the ligand might remain bound with MOR other for a longer period.
Morphine- the MOR-Morphine complex showed in its 4th run with the lowest binding energy being -8.05. The 2D image of the complex indicates one conventional H-bond with LEU:192 and 2 conventional H-bonds with PHE:234. It shows 2 pi-alkyl bonds with PRO:326 and one alkyl bond with PRO:194. The 3D image showed the medium aromaticity, low H bonds, and neutral charge. They are non-polar too.
Noscapine- The lowest binding energy was found at -7.86 at its 4th run. The 2D image reveals two conventional bonds with PHE:234 and 2 pi alkyl bonds with PRO:236 and PRO:194; and one alkyl bond with CYS:233. The 3D analyses show medium aromaticity, low hydrogen bonds, and neutral charge resulting in a non-polar structure.
Papaverine- the lowest binding energy of the MOR and papaverine complex was found to be -8.15 at the 5th run. The 2D structure showed (i) conventional H Bonds (ii)Carbon Hydrogen bond (iii) alkyl bond (iv) pi-alkyl (v) pi-lone pair (vi) pi-sigma pair with the aminoacids PRO:236, PRO:194, THR:321, VAL:320, LYS:280, PHE:278, SER:191, CYS:233, PHE:234, ARG:150, and LEU:192. The 3D structure showed high aromaticity indicating highly non-polar; low hydrogen bonds and neutral charge. As compared to other complexes this complex has a slight positive charge in its structure and shows less negative charge in them.
Thebaine- The MOR-thebaine complex had its lowest binding energy of -8.17 shown at its 5th run. The 2D structure showed carbon-hydrogen bonds involving THR:321 and ASP322 and conventional hydrogen bonds with PHE:278 and PHE:234. It also showed alkyl and pi-alkyl bonds with PRO:236, PRO:194, and CYS:233. The 3d structure of the complex showed a low level of aromaticity indicating less non-polarity as compared to the other natural opiates. The model shows low hydrogen bonds and a neutral charge.
The analysis for synthetic opioids is as follows:
Buprenorphine- The MOR-buprenorphine complex had its lowest binding energy of -9.10 was shown at its 6th run. The 2D structure showed conventional hydrogen bonds associated with amino acids LEU:192 and ALA:193; carbon-hydrogen bonds with amino acids THR:321, and SER:191; pi-anion with ASP:322, and alkyl and pi-alkyl bonds with VAL:320, PRO:236, PRO:194 and CYS:233. The 3d structure of the complex showed a high level of aromaticity indicating high non-polarity. The model shows moderate-level hydrogen bonds and a neutral charge.
Carfentanyl- The lowest binding energy of the MOR and carfentanyl complex was found to be -7.02 at the 9th run. In the 2D structure, four hydrogen bonds can be seen with VAL:320, ASP:322, THR:321, and five alkyl and pi-alkyl bonds with PRO:236, PRO:194, PHE:278, CYS:233. The 3d structure shows a very high presence of aromatic rings implying very high non-polarity. However, the hydrogen bond present is very low and the map of charges shows both the presence of positive and negative charges. Unlike other complexes, this complex has a positive charge on its left side.
Fentanyl- the lowest binding energy of the MOR and fentanyl complex was found to be -7.14 at the 7th run. The 2D structure showed three hydrogen bonds with the amino acids THR:321, VAL:320, and HIS:62; and alkyl and pi-alkyl bonds with PRO:236, ARG:150, and LEU192. The 3D complex of the MOR with fentanyl complex also shows a very high presence of aromaticity thus, a very high non-polar nature. Further analysis showed high hydrogen bonds and neutral charge.
Hydrocodone- the lowest binding energy of the MOR and hydrocodone complex was found to be -8.19 at the 5th run. The 2D structure of the complex showed two hydrogen bonds at PHE:278, and PHE:234 and pi-alkyl and alkyl bonds at PRO:194, PRO:236, and CYS:233. The 3D docking model showed a moderate presence of aromaticity indicating a non-polar nature, low presence of hydrogen bonds, and neutral charge.
Hydromorphone- the lowest binding energy of the MOR and hydromorphone complex was found to be -8.39 at the 2nd run. The 2D showed two hydrogen bonds with PHE:278 and LEU:192 and pi-alkyl bonds with PRO:194 and PRO:236. The 3D interaction in Discovery Studio showed low aromaticity indicating non-polarity and hydrogen bonds with a neutral charge.
Levomethadone- the lowest binding energy of the MOR and levomethadone complex was found to be -6.85 at the 3rd run. The 2D structure showed hydrogen bonds with PHE:278, VAL:320, and THR:321 and pi-alkyl bonds with CYS:233 and PRO:194. The 3D structure showed a high presence of aromaticity gain indicating a non-polar nature, moderate amount of hydrogen bonds, and neutral charge. In terms of charge, this complex shows a slightly more positive charge in its upper left.
Methadone- the lowest binding energy of the MOR and methadone complex was found to be -6.40 at the 9th run. The 2D structure showed two hydrogen bonds with ASP:322 and PRO:236 and pi-anion bond with ASP:322 and pi-sulfur bond with CYS:233 and three pi-alkyl bonds associated with LYS:280, and PRO:236. The 3D structure showed moderate aromaticity further indicating, low hydrogen bonds, and neutral charge.
Oxycodone- the lowest binding energy of the MOR and Oxycodone complex was found to be -8.27 at the 4th run. The 2D structure of the complex showed three hydrogen bonds with THR:321, PHE:278, and PHE:234 and pi-alkyl and alkyl bonds with PRO:194, PRO:236, and CYS:233. The 3D model of the MOR-oxycodone complex showed moderate aromaticity, moderate levels of hydrogen bonds, and neutral charge. It also has a non-polar nature
Oxymorphone- the lowest binding energy of the MOR and Oxymorphone complex was found to be -8.39 at the 9th run. The 2D structure showed three hydrogen bonds with THR:321, LEU:192, and PHE:278; and pi-alkyl bonds with PRO:236 and PRO:194. The 3D structure shows moderate level aromaticity and hydrogen bonds with the neutral charge. It is non-polar in nature.
Pethidine- the lowest binding energy of the MOR and Pethidine complex was found to be -6.78 at the 7th run. MOR-pethidine is the only 2D structure that showed unfavorable bond formation at PHE:234. However, there are other bonds present which are hydrogen and alkyl bonds that are associated with THR:321, ASP:322 and PRO:236, CYS:233 respectively. The 3D structure showed low aromaticity, hydrogen bonds, and neutral charge. It is also non-polar in nature.
Remifentanil- the lowest binding energy of the MOR and Remifentanil complex was found to be -6.21 at the 7th run. The 2D structure of the remifentanyl with MOR showed a much more complex structure as compared to other complexes. The bond formations were hydrogen bonds with ARG:150, SER:191, SER:277 PHE:234 and alkyl and pi-alkyl bonds with LEU:192,HIS:62, PRO:194, PRO:236, LYS:280, PHE:278 and CYS:233. The 3D structure of MOR with remifentanil showed high aromaticity implying a non-polar nature, moderate hydrogen bonds, and neutral charge.
Tapentadol- the lowest binding energy of the MOR and Tapentadol complex was found to be -6.15 at the 9th run. The 2D structure showed hydrogen bonds with THR:321, ASP:322, and LEU:192; alkyl and alkyl and pi-alkyl bonds with VAK:320, PRO:236 and PHE:278. This structure showed further a pi-lone pair with PHE:234. The 3D structure showed a moderate level of aromaticity, low hydrogen bonds, and a neutral charge. It is also analyzed to be hydrophobic in nature.
Tramadol- the lowest binding energy of the MOR and Tramadol complex was found to be -6.99 at the 5th run. The 2D structure of the complex shows hydrogen bonds with LEU:192, PHE:235, ASP:322, PHE:234, and PHE:278; and alkyl and pi-alkyl bonds with CYS:233, PRO:236, LYS:280, and VAL:320. The 3D structure showed low aromaticity, hydrogen bonds, and neutral charge (with less negative charge). Tramadol is also hydrophobic in nature.
ADME Analysis
The Boiled Egg representation of the ADME result shows every ligand except for Noscapine, Remifentanil, Oxycodone, and Oxymorphone penetrates through the blood-brain barrier (white). Meanwhile, these four are absorbed by the intestines (yellow) (Figure 7).

Conclusion
This research was done on a total of 18 natural and synthetic opioids. By the computational technique of molecular docking, these opioid drugs were docked with the MOR of Homo sapiens to study the MOR-ligand complex. Molecular Docking is a very helpful method for the studies of drugs and to predict the structure of the complex. These studies are especially utilized in drug discovery. among these eighteen drugs Buprenorphine, Hydromorphone, and Oxymorphone with the lowest binding energy of -9.10, -8.39, and -8.39 respectively. The ADME analysis done with the opioid drugs played a major role in analyzing the penetrability of the drugs through the blood-brain barrier and their absorbance by the intestine. Oxycodone, Oxymorphone, Remifentanil, and Noscapine were found to be well absorbed by the intestines. And the others, Codeine, Morphine, Papaverine, Thebaine, Buprenorphine, Carfentanyl, Fentanyl, Hydrocodone, Hydromorphone, Levomethadone, Methadone, Pethidine, Tapentadol, Tramadol easily could pass the blood-brain-barrier according to the study. Molecular Docking and ADME analysis are two essential techniques that can help in the research field of toxicology.